产品中心

X射线衍射(XRD)是所有物质,包括从流体、粉末到完整晶体,重要的无损分析工具。对材料学、物理学、化学、地质、环境、纳米材料、生物等领域来说,X射线衍射仪都是物质结构表征,以性能为导向研制与开发新材料, 宏观表象转移至微观认识,建立新理论和质量控制不可缺少的方法。通过对置于分光器(测角仪)中心的样品上照射X射线,X射线在样品上产生衍射,改变X射线对样品的入射角度和衍射角度的同时,检测并记录X射线的强度,可以得到X射线衍射谱图。用计算机解析谱图中峰的位置和强度关系,可以进行物质的定性分析、晶格常数的确定和应力分析等。而且通过峰高和峰面积也可进行定量分析。除此以外,通过峰角度的扩大或峰形进行粒径、结晶度、精密X射线结构解析等各种分析,还可以进行高低温及不同气氛与压力下的结构变化的动态分析等。
常用于测定无机物、有机物及配位化合物(晶体状态)分子的准确三维立体结构,通过晶体结构计算出测定分子详细的键长、键角、 构型、构象、成键电子密度及分子在晶格中的排列情况,是对化合物直接、可靠的鉴定。
XRD-科研测试站
常规XRD:
1.样品状态:可为粉末、块状、薄膜样品、液体样品可以涂在载玻片上干燥之后测试(有可能测试到基底,请悉知)。
2.粉末样品:颗粒一般不超过75微米,手摸无颗粒感,较大需要适当研磨,样品用量一般0.5g以上,(如样品密度偏大,请适量增加),粒度<200目,越细越好。
3.块状、薄膜样品:a.至少有一表面为平整光洁平面。b.若为立方体则平面长宽≤20mm,且厚度范围50微米~15毫米;c.若为圆柱体则直径≤20mm,高≤15mm;一定要标明测试面!
注意:1.请尽量用样品管送粉末样,用样品袋盛装难以刮下且易损失,可能影响测试效果。
2.较薄的薄膜/涂层样品(厚度<100nm)请慎重选择广角XRD;
3.微区XRD样品需标记待测点,束斑最小为0.01mm,样品高度应不超过8mm,待测面应平整光滑。
单晶XRD:
1. 晶体质量好,可测无机晶体,有机晶体以及MOF
2. 有机和MOF类单晶,在送样时,应放在母液中,一起寄出
3. 若要解析结构,应提供分子式以及结构式
掠入射X射线衍射(GIXRD):
1.整体长宽1cm;
2.镀层或者薄膜一般厚度不要低于50nm,否则可能测不出信号;
3.粉末、液体等其他无法测试。
1.为什么要求XRD测试粉末样品要稍微多一些?
因为粉末样品要铺满整个样品台,不然X射线可能会打在样品台上,影响数据质量。
2.为什么XRD测试要求薄膜(块体)样品尺寸要合适?
因为放置薄膜(块体)样品的样品台尺寸是固定的,用橡皮泥来固定样品,样品太大放不进去,样品太小不好固定。
3.为什么XRD数据的峰强度较低,甚至没有明显的衍射峰?
样品的衍射峰强度最主要跟样品本身的结晶度有关,其次跟样品量以及仪器的功率都有关系。
4.粉末样品量过少会有什么样的影响?
因为粉末样品要铺满整个样品台,样品量过少可能导致X射线打在样品台上,从而影响数据质量。
5.为什么部分样品的测试结果中衍射峰强度较低,甚至没有明显的衍射峰?
样品的衍射峰强度主要跟自身结晶度有关,结晶度越好,衍射峰越明显,其次才是样品量和仪器功率。
6.步长和扫描速度有何关联?扫描速度对样品的测量有何影响?
步长决定了扫描的精细程度,扫描速度是由步长和每步的时间决定的,每步的时间长短会影响衍射峰的强度和信噪比。每步的时间越长,步长越小,则谱越精细,信噪比越高,可用于定量计算,如XRD精修等。
7.XRD定量的精确度如何?
XRD为半定量分析,定量测试结果精确度有限,仅供参考。
8.为何有些样品的测试结果基线较高?能否改善?
有荧光散射现象的样品,如铁含量较高的样品,测试结果会出现基线较高的情况,这是无法改善的。
9.Cu靶的波长是多少?
Kα1=1.540538埃。
10.XRD掠入射角度如何选择?
掠射角度建议越小越好,一般都是0-1°最好,例如0.5°,不建议2°或3°,这种基本不叫掠射,基底的信号也会比较明显
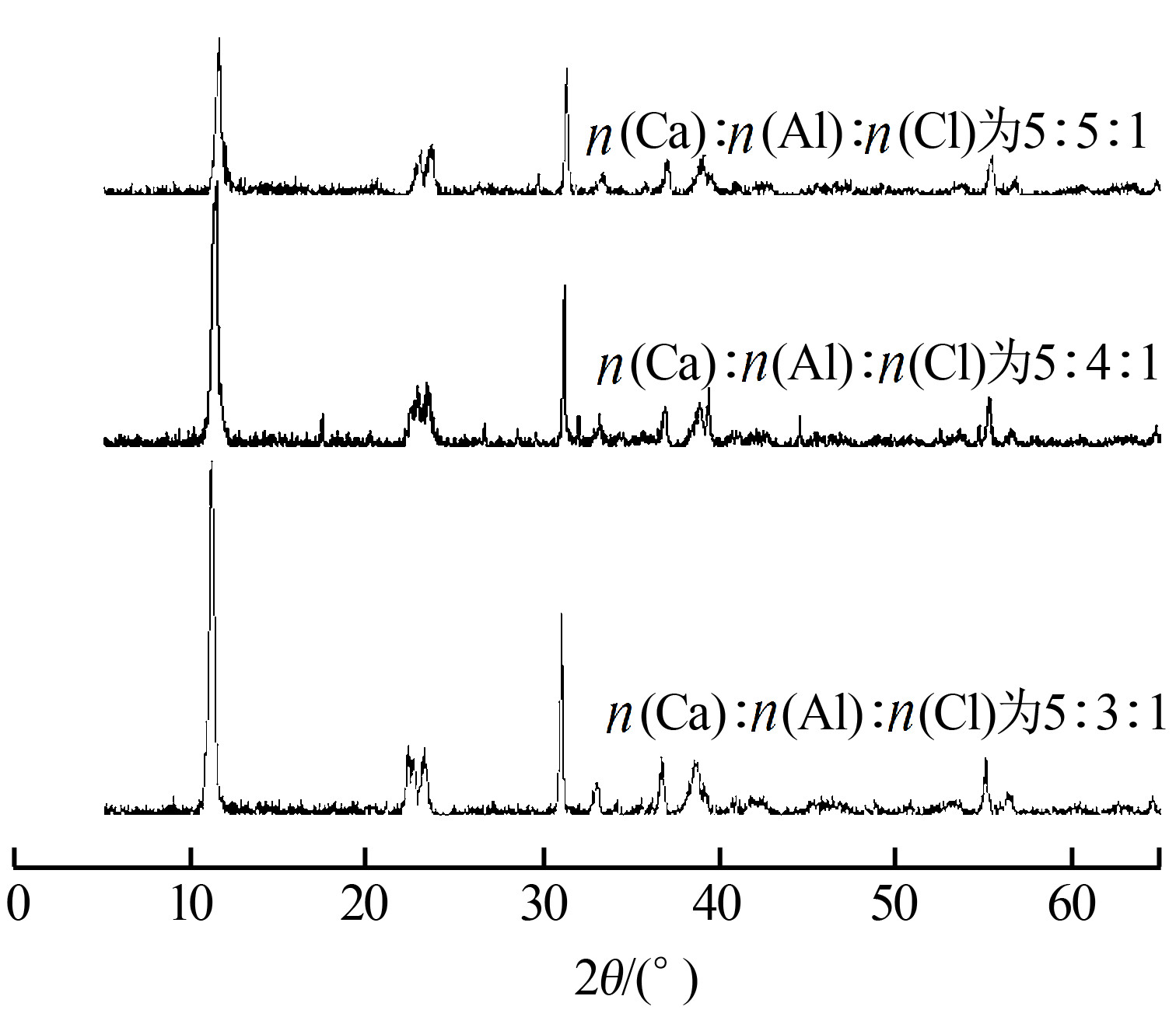
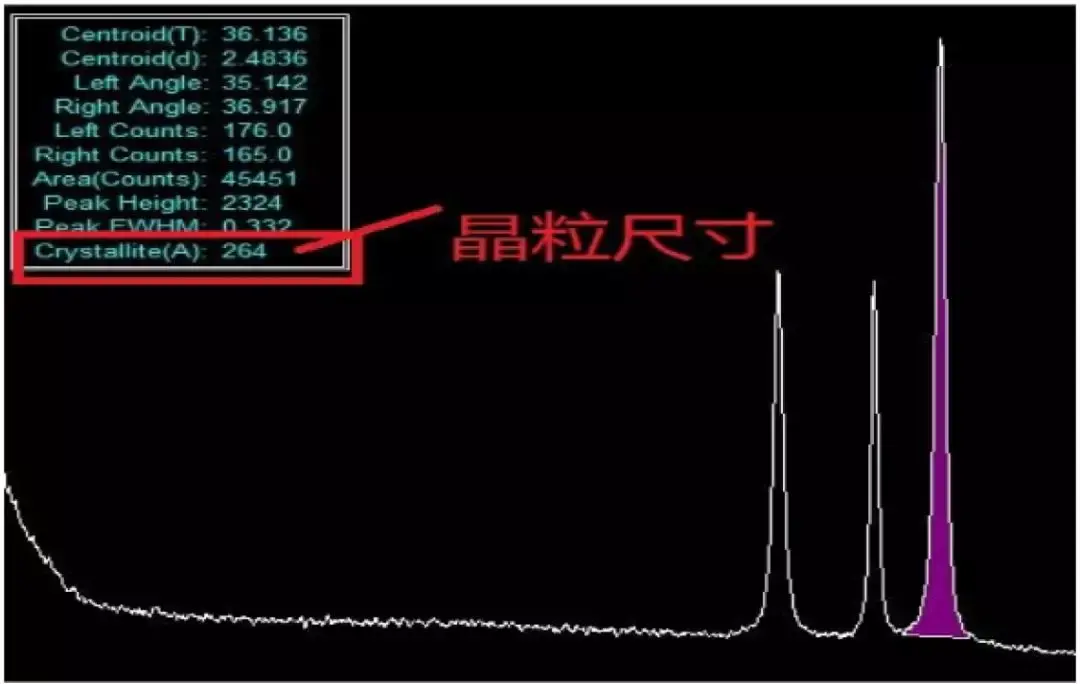
11.XRD如何计算晶粒尺寸?
XRD计算晶粒尺寸需要知道峰的归属,如果可以对上标准谱,可以用卡片里的晶面指数代替,如果完全未知,需要先对谱图指标化,指标化的过程比较麻烦。
12.XRD小角掠入射测试深度是多少?
深度是一方面根据入射角来定的,另一方面还由样品的元素决定,入射深度可以进行计算,但是比较麻烦,元素的吸收系数都得考虑,通常情况下简单通过入射角可以粗略确定,如果要准确计算,需要查阅相关的文献资料。
13.薄膜状样品做XRD小角掠射只出来基底峰是什么原因?
一般膜层越薄,掠射角选取越小,一般是0.5-2度,理论上出基底峰的话也会有膜层的峰,但是如果膜层很薄只有几十纳米的话,即使掠射角做到0.5,出膜层衍射峰的几率也不大,即使出来也会被强大的基底峰掩盖住。
14.X射线单晶衍射仪和x射线多晶衍射仪有什么区别?
1)单晶衍射仪主要用于测定单个纯物质的晶体结构,对于已知结构,可以进行精修,对于未知结构,可以鉴定结构。要求所测的样品为块状单晶。一般在表征新化合物时,最好用单晶衍射仪,测量一个单晶体需要一到两天,解析一个单晶可能要花费更多的时间。另外,培养单晶也很不容易,单晶生长受很多条件的限制。2)多晶体衍射仪(XRD)也称为粉晶衍射,主要是用来测定样品的物相组成,它主要依据的PDF数据库,通过查找这个库中与样品衍射谱相同的物相来鉴定某个物相是否存在,因此,鉴定的必须是已知物相。也可以测量单晶,前提条件是把单晶破碎成粉晶,这时测量的相当于是纯物质。对于样品要求块状、粉末状都可以,样品容易制取,测量时间短,物相鉴定现对来说比较简单、快速。
15.XRD测试基线很飘的原因?
XRD测试数据基线飘是正常的,仪器光斑开的比较大,检测器窗口也大,这样的图谱信噪比和强度都会好很多,所以起始角度越低,前面的光子计数就越高。对数据分析没什么影响,不用刻意进行处理。
16.XRD小角测试应当注意的问题?
(1)如果看10度以下的峰,需要确认测试目的是看物相还是看孔结构;
(2)如果看物相,需要用广角模式测试,也就是测试5-90(或者3-90度都行),如果看孔结构,需要用小角模式,也就是0.5-10度;
也就是说,如果都是测试5-10度,用小角模式和广角模式,结果是不同的。
17.高温XRD随着温度升高,峰位为什么会出现左移现象?
随温度升高使衍射峰左移,这是因为一般材料都是正膨胀系数材料,即随温度升高而膨胀。根据布拉格公式2dsinθ= nλ,在波长不变的情况下,d值增大,必然使sinθ变小,而在衍射范围内,也就是θ变小,即峰位左移。



 科研测试站
科研测试站





